Integration of summary data from GWAS and eQTL studies
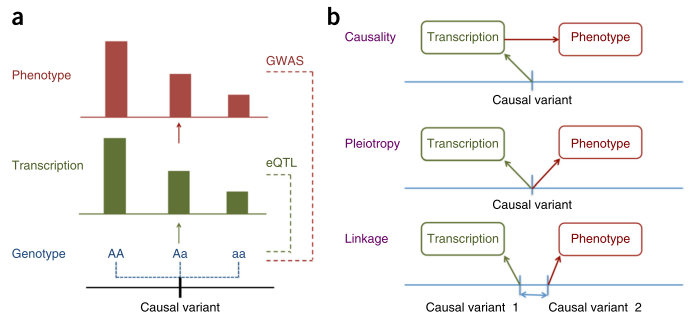
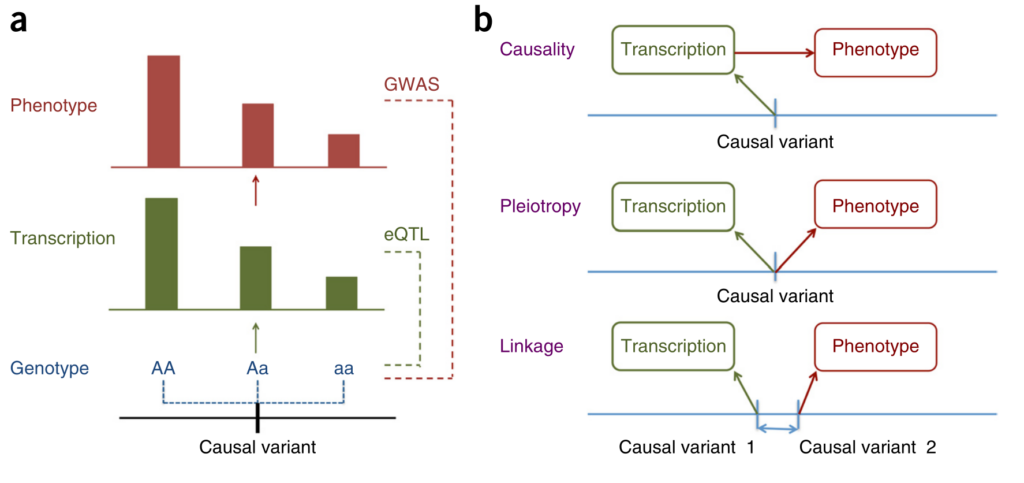
Figure 1 from [1].
Zhu and colleagues approach this question from the perspective of Mendelian Randomisation (MR). They show that it is not possible to distinguish between causation and pleiotropy — at least not by using the data from a single variant and two traits [2]. Probably the most interesting methodological result of this paper is the HEIDI (heterogeneity in dependent instruments) test, which, essentially, combines the results from two GWAS and knowledge about the Linkage Disequilibrium (LD) structure to address the question whether the pattern of associations observed across the region is sufficiently similar to hypothesise presence of a single underlying functional variant which affects both traits (either through causation or pleiotropy). The idea of the test is explained in Figure 1 of the manuscript. The group implemented the methods they have developed in the SMR software [3].
The authors have applied the developed methodology to study loci generated in GWAS of five complex traits, by exploiting the results from a large eQTL study [4] performed on peripheral blood [5]. Very interestingly, from 101 of the annotated gene-probes which passed the HEIDI test, 2/3 (62) did not locate in the gene nearest to the strongest complex trait association signal. All of us geneticists do know that it is plausible to have a variant which regulates a gene located as far as few Mb apart, but it is very good to have this quantitative assessment.
One question which, in our view, is only partly addressed in the paper is the procedure to select probes to be tested by HEIDI with the trait of interest and when to claim ‘no significant deviation’ [6]; what threshold values of the test(s) should be used when claiming possibility of a shared functional allele between the complex and transcription traits [7]? What about the choice of SMR-test SNP? (in this work the eQTL SNP goes into SMR, which is kind of logical in the context of MR; but generally speaking it may be not so obvious)
The fact that the authors present their work in the context of the investigation of a complex trait locus by aligning it with results from an eQTL study should not confuse the reader; in principle the method is broader and could — at least in our view — be applied to any pair of traits.
Altogether – this is a great work, and we highly recommend to read it carefully. We at PolyOmica look very much forward to trying this method on our favourite loci underlying complex and omics traits.
[1] Zhu et al., Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. doi:10.1038/ng.3538
[2] In no way does the latter makes this paper weaker; such a result is actually expected in the context of MR research
[3] SMR software, http://cnsgenomics.com/software/smr/
[4] Westra et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. doi:10.1038/ng.2756
[5] Yes, we all know this is not ideal; but it is handy and may be reasonable given large overlap of eQTLs between tissues (see results of GTEx). UPDATE: click HERE to see extended discussion
[6] This is somewhat against our usual ways: normally we are happy when we have low p-values. Here we should be happy with high p-values. So this is like testing Hardy-Weinberg Equilibrium. E.g. in GWAS QC many people use p-HWE > 1e-6, but this is quite arbitrary.
[7] May be one could better approach such type of questions via Bayesian perspective. Fairly, we do not know (only yet).
No Comments